
1. INTRODUCTION
Wood is an abundant biomass consisting of cellulose, hemicellulose, and lignin. Hemicellulose is a heteropolymer of plant cell walls and the second most abundant polysaccharide on the earth after cellulose. The average content of hemicellulose in plant cell walls is 30% (Cheng, 2014). Therefore, an efficient usage of hemicellulose is essential to make plant resources economically feasible.
Xylanase (EC 3.2.1.8) degrades the polysaccharide chain present in xylan, which is a component of hemicellulose (Cheng, 2014). A wide range of microorganisms produce xylanases, such as Bacillus sp. (Sá-Pereira et al., 2004), Enterobacter sp. (Khandeparkar and Bhosle, 2006), Streptomyces sp.(Ninawe et al., 2008), and Thermomyces lanuginosus (Singh et al., 2003). Microbial xylanases are commonly used in the pulp–paper industry for biobleaching and in the food and drink industry for improving flavor, texture, aroma, and digestibility (Beg et al., 2001; Kim and Paik, 1997; Kim et al., 2007; Polizeli et al., 2005). In addition, they are used in the bioenergy industry for bioconversion of lignocellulosic biomass. They can be used with cellulases as accessory enzymes to increase the overall fermentable sugar yields (Rio et al., 2010).
Our laboratory has been collecting wood decaying fungi for wood preservation studies. The wood decay of all the collected 56 fungi was evaluated. Among them, Acanthophysium sp. KMF001 (KCTC No. 18282P) exhibited a high cellulase activity (Shin et al., 2016) and a high wood decaying activity (Kim et al., 2016). The Acanthophysium genus is a ubiquitous fungi and Acanthophysium lividocaeruleum, a wood decaying fungi, is the well-known species in this genus (Chedgy et al., 2009). However, the high wood decaying activities of species in the Acanthophysium genus have not been well studied.
When the extracellular crude enzymes of Acanthophysium sp. KMF001 were applied in biopolishing of cotton fabric, the effectiveness of the enzymes was comparable to commercial enzymes (Shin et al., 2016). The saccharification activity of the extracellular crude enzymes of Acanthophysium sp. KMF001 was also strong enough to produce glucose from woods as much as a commercial enzyme (Kim et al., 2016). Because of the wide spectrum of chemicals in woods, the wood decaying process requires a complex enzymatic reaction. In consideration of an efficient wood decay, Acanthophysium sp. KMF001 might have a wide range of enzymes including xylanases. Because enzymes are the biological tools for utilization of biomass, identifying novel enzymes will enrich the knowledge of the reaction mechanism, the design of new enzymes, and their industrial utilization.
In this study, a novel glycoside hydrolase gene from Acanthophysium sp. was identified through transcriptomic analysis. The protein encoded by this novel gene was expressed heterogeneously in Escherichia coli BL21 (DE3) which exhibited the activity of xylanase. The enzymatic properties of the expressed new xylanase were characterized in this study.
2. MATERIALS and METHODS
Acanthophysium sp. KMF001 (KCTC No. 18282P) was cultured on potato dextrose agar (PDA; Becton, Dickinson and Company, Franklin Lakes, NJ, USA) plates. For the liquid culture, potato dextrose broth (PDB; Becton, Dickinson and Company) or TYE-cellulose media (7 g/l tryptone, 3 g/l yeast extract, 5 g/l KH2PO4, 5 g/l K2HPO4, 3 g/l MgSO4·7H2O, and 20 g/l cellulose) was used and cells were incubated at 26°C.
E. coli BL21(DE3) and E. coli competent cells (One Shot™ TOP 10, Thermo Fischer Scientific Korea, Seoul, Korea) were cultured aerobically at 37°C in Luria-Bertani medium (LB; Becton, Dickinson and Company) containing 50 μg/ml of kanamycin. E. coli competent cells and E. coli BL21(DE3) were used as the cloning hosts and the expression host, respectively. Plasmid pET-24a(+) was used as the expression vector.
For total RNA isolation, Acanthophysium sp. KMF001 was cultured in PDB and TYE-cellulose media independently at 26°C for 5 days. Each culture was used for total RNA isolation separately and the isolated total RNAs of both were combined and used for the purification of mRNA. Total RNA was isolated according to a previous study (Rio et al., 2010) using TissueLyser LT (Qiagen Korea Ltd., Seoul, Korea) and TRIzol®RNA (Thermo Fisher Scientific Korea). The quality and the quantity of extracted RNA were determined by measuring the absorbance at 260 nm and 280 nm using UV spectrophotometer (Optizen 2120UV, Mecasys Co., Ltd, Daejon, Korea) and calculating their ratio.
A process for separating mRNA from total RNA was performed according to the manual of PolyATract® mRNA Isolation System (Promega Co., Madison, WI, USA). Transcriptomic analysis was performed by Macrogen Korea (Seoul, Korea) using GS-FLX Titanium (Roche Diagnostics Korea Co., Ltd, Seoul, Korea). The sequence analysis was processed by GS De Novo Assembler (Roche Diagnostics Korea Co., Ltd) using a transcriptome shotgun assembly method. Isotig sequences were made by connecting two or more singletons that exist in the same gene regions using the GS De Novo Assembler.
Among the total isotig sequences, xylanase candidate genes were selected based on the suggested biological function from the tblastn search in the translated nucleotide database of the National Center for Biotechnology Information (http://blast.ncbi.nlm.nih.gov/Blast.cgi). Among them, isotig01043 xylanase candidate genes were selected for the expression in E. coli based on the similarity and coverage of a suggested open reading frame (ORF) when it compared with the most similar polypeptide sequence among the search results.
The independently separated total RNA from PDB or TYE-cellulose media was combined and used as the template for cDNA synthesis of isotig01043 using M-MLV Reverse Transcriptase (GenDEPOT Co., Barker, TX, USA) according to its manual. The cDNA was stored at −80°C and used as the template for amplification.
For the amplification of isotig01043 xylanase gene, polymerase chain reaction was used in two consecutive rounds. In the first round, isotig01043 gene containing the untranslated region was amplified using primers with a high sequence specificity and in the second round, the isotig01043 gene was amplified using the first-round amplified product as the template and another set of primers with designed restriction enzyme sites. The pair of primers for the first-round amplification were 01043F-out (5′-TGATTGCTGTCCTTCACTCTCA-3′) and 01043R (5′-CAAGCCCAAAAGGTAGATGG-3′). The pair of primers for the second-round amplification were NdeI_24_F (5′-AAGGAGATATACATATGATT GCTGTCCTTCACTCTCATTC-3′) and XhoI_24_R (5′ -GGTGGTGGTGCTCGAGCAGCATCCCAATGATC ATAC-3′). The restriction enzyme sites of NdeI and XhoI in NdeI_24_F and XhoI_24_R primers, respectively, are underlined. SP-Taq DNA polymerase (LaboPass™ CMT2001, Hokkaido System Science Co., Ltd, Sapporo, Japan) was used as the DNA polymerase.
The expression vector, pET-24a(+), and the amplified isotig01043 gene were cut using both NdeI and XhoI restriction enzymes (New England Biolabs™ Inc., Beverly, MA, USA) to insert the amplified new xylanase product from cDNA. T4 ligase (Promega Co.) was used for ligation according to its manual. The recombinant plasmid was transformed into E. coli competent cells using One Shot™ TOP10 by the heat shock method according to its manual. A total of 50 μl of transformed cells was spread on the pre-warmed LB agar plate containing 50 μg/ml kanamycin and incubated at 37°C for 1 day. Sixteen single colonies grown on plates were randomly selected and the presence of the correct recombinant plasmid was checked by polymerase chain reaction using a pair of primers, T7 (5′-TAATACGA CTCACTATAGGG-3′) and T7 terminator (5′-GCTA GTTATTGCTCAGCGG-3′). To confirm the presence of the new xylanase gene in the recombinant plasmid, the confirmed colonies were cultured in LB media and their plasmids were purified using QIAprep Spin Miniprep Kit (Qiagen Korea, Ltd.). The inserted gene was sequenced by Cosmogenetech Co., Ltd. (Seoul, Korea). The constructed recombinant plasmid, pET-24a (+) containing the new xylanase gene, was transformed into E. coli BL21(DE3) using the heat shock method again. The successfully transformed E. coli BL21(DE3) were selected by culturing on LB agar plates containing 50 μg/ml kanamycin after incubation at 37°C overnight.
The solubility of the expressed protein was tested according to the QIAexpressionist™ handbook (Qiagen Korea Ltd.). The protein encoded by the new xylanase gene was expressed from the transformed E. coli BL21(DE3) according to the QIAexpressionist™ handbook (Qiagen Korea Ltd.). The expressed xylanase was present in the inclusion bodies of cells. The expressed protein was used in the protein refolding procedure according to a previous study (Burgess, 1996) with few modifications. When the proteins were refolded with dilution after dissolving with 6 M guanidinium chloride, there was an additional 15-min pause for every two-fold dilution, and the refolded proteins were filtered using a 0.45-μm membrane filter (Merck KGaA, Darmstadt, Germany) for the final clarification, rather than 0.22-μm membrane filter.
The protein concentration was measured using the Bradford’s method with bovine serum albumin as a standard (Bradford, 1976) according to the manufacturer’s manual (Bio-Rad Laboratories, Inc., Hercules, CA, USA).
The new xylanase was purified using a 5-ml polypropylene column (Qiagen Korea Ltd.) packed with 0.5 ml of Ni-NTA agarose resin (Qiagen Korea Ltd.). The packed column was equilibrated with lysis buffer, loaded with protein samples, and washed four times with 1 ml of wash buffer (50 mM NaH2PO4, 300 mM NaCl, and 20 mM imidazole at pH 8.0). The remaining xylanase proteins were eluted four times with 0.5 ml of elution buffer (50 mM NaH2PO4, 300 mM NaCl, and 250 mM imidazole at pH 8.0). The purity of each elute was analyzed by sodium dodecyl sulfate– polyacrylamide gel electrophoresis, as described in the following section.
Sodium dodecyl sulfate-polyacrylamide gel electrophoresis was performed using a 12% polyacrylamide gel (Laemmli, 1970). Following electrophoresis, the separated proteins were stained with staining buffer (1 g/l Coomassie brilliant blue R-250, 450 ml methanol, 100 ml glacial acetic acid, and distilled water up to 1 l) for 30 min, and the gels were visualized using Gel Doc™ XR+ Imager (Bio-Medical Science Co., Ltd, Seoul, Korea). Xpert 2 Prestained Protein Marker (GenDEPOT Co.) was used as the size marker.
The following enzyme activities were measured according to previous studies: endo-β-1,4-glucanase (Quevedo-Hidalgo et al., 2012), cellobiohydrolase (Quevedo-Hidalgo et al., 2012), β-glucosidase (Joo et al., 2009), exo-1,3-1,4-glucanase (Nakatani et al., 2010), xylan-1,4-β-xylosidase (Choengpanya et al., 2015), chitosanase (Piza et al., 1999), and endo-β-1,4-xylanase (Guo et al., 2009).
The effect of pH on the enzyme activity was measured at 30°C. The buffers used for pH adjustment were 100 mM sodium citrate buffer in the pH range of 3.0–4.0, 100 mM sodium acetate buffer in the pH range of 4.0–6.0, 100 mM sodium phosphate buffer in the pH range of 6.0–8.0, and 100 mM Tris-HCl buffer in the pH range of 8.0–9.0. The effect of temperature on the enzyme activity was measured using 100 mM sodium acetate buffer (pH 5.0) over the temperature range of 20–70°C with 10°C intervals. The thermal stability of the enzyme was measured over the temperature range of 10–40°C with 10°C intervals up to 4 h. After incubation, the xylanase activity was measured at pH 5.0 and 30°C.
For the effect of various ions on the xylanase activity, 11 ion compounds, CuSO4, CaCl2, CoCl2, KCl, BaCl2, FeSO4, ZnSO4, MnCl2, MgCl2, FeCl3, and NaCl at 1 mM concentration, were added into the enzyme solution. The enzyme activity without any additional ion was used as the control activity, and the relative activity change was calculated based on this control.
To analyze the kinetic properties of the purified new xylanase, the reaction rate was analyzed at three concentrations of beech wood xylan, 5, 10, and 20 mg/ml. The experimental results were plotted to determine the Michaelis–Menten constant (Km) and the maximum reaction velocity (Vmax) according to the Lineweaver–Burk plot (Lineweaver and Burk, 1934).
3. RESULTS and DISCUSSION
The extracellular crude enzyme of Acanthophysium sp. KMF001 exhibited strong cellulase (Shin et al., 2016) and wood decaying activities (Kim et al., 2016). The transcriptomic analysis identified 1886 assembled genes by assembling singleton fragments. Among them, 1148 assembled genes exhibited their biological function by gene ontology analysis. Among the 1148 genes, only 17 genes had more than 90% identity with its most similar gene in the NCBI database. This low number of genes with more than 90% identity suggested that Acanthophysium sp. KMF001 was a novel strain and its genetic information was unveiled. All the five genes in Table 1 had less than 90% identity, as well. These comparison results shown in Table 1 suggested the uniqueness of putative glycoside hydrolases in Acanthophysium sp. KMF001.
To identify xylanase in Acanthophysium sp. KMF001, the total mRNAs of cells were isolated and analyzed using the next-generation sequencing method (unpublished data). The sequencing result revealed 14 candidate genes for glycoside hydrolases (Supplementary Figs. S1 and S2). Among them, only five sequences had a complete ORF based on the comparison with the homologous sequences. The proteins that have the highest similarity with the ORF of each gene in the NCBI non-redundant protein sequences database using the blastp search are listed in Table 1. Among the five candidate genes, isotig01043 was selected for heterogeneous expression in this study.
The identity of the novel protein encoded by isotig01043 was 83% (Table 1), when compared with the closest protein in the NCBI database, a hypothetical protein from Stereum hirsutum. This low identity did not suggest the biological activity of the novel protein, but it had a conserved domain of glycosyl hydrolase family 16 (GH16). The protein encoded by isotig01043 had a domain similar to the GH16_fungal_Lam16A_ glucanase domain specifically (Fig. 1), instead of the GH_XET domain, which is the only domain hydrolyzing the xylose polymer in GH16. Therefore, the endo-β-1,4-xylanase of isotig01043 was a new type of enzyme with a new domain.

To identify the introns, the region of isotig01043 on the chromosomal DNA was amplified by polymerase chain reaction, sequenced, and aligned with isotig01043, as shown in Supplementary Fig. S3. This alignment showed the existence of nine introns in the region of isotig01043 on the chromosomal DNA.
Isotig01043 was introduced in the protein expression vector, pET-24a(+), (Supplementary Fig. S4) and transformed into E. coli BL21(DE3). The 45 bases in front of the start codon were included because of uncertainty of ORF based on a low query coverage, shown in Table 1. The sequencing results after the construction of the recombinant DNA with pET-24a(+) confirmed the isotig01043 gene. The recombinant isotig01043 was expressed with 1 mM isopropyl β -D-1-thiogalactopyranoside. All expressed proteins were found in the inclusion bodies of cells. The insoluble expressed proteins were solubilized by 6 M guanidinium chloride. The solubilized and denatured proteins were renatured by dilution. The renatured isotig01043 was purified with a Ni-NTA column (Qiagen Korea Ltd.). Because the type of glycoside hydrolase of the purified proteins was not yet identified, various glycoside hydrolase activities of the purified proteins were measured (Table 2). Among the seven different enzymatic activity assays, only endo-β-1,4-xylanase activity was detectable at 0.235 U/ml. This result suggested that the expressed protein from isotig01043 was endo-β-1,4-xylanase.
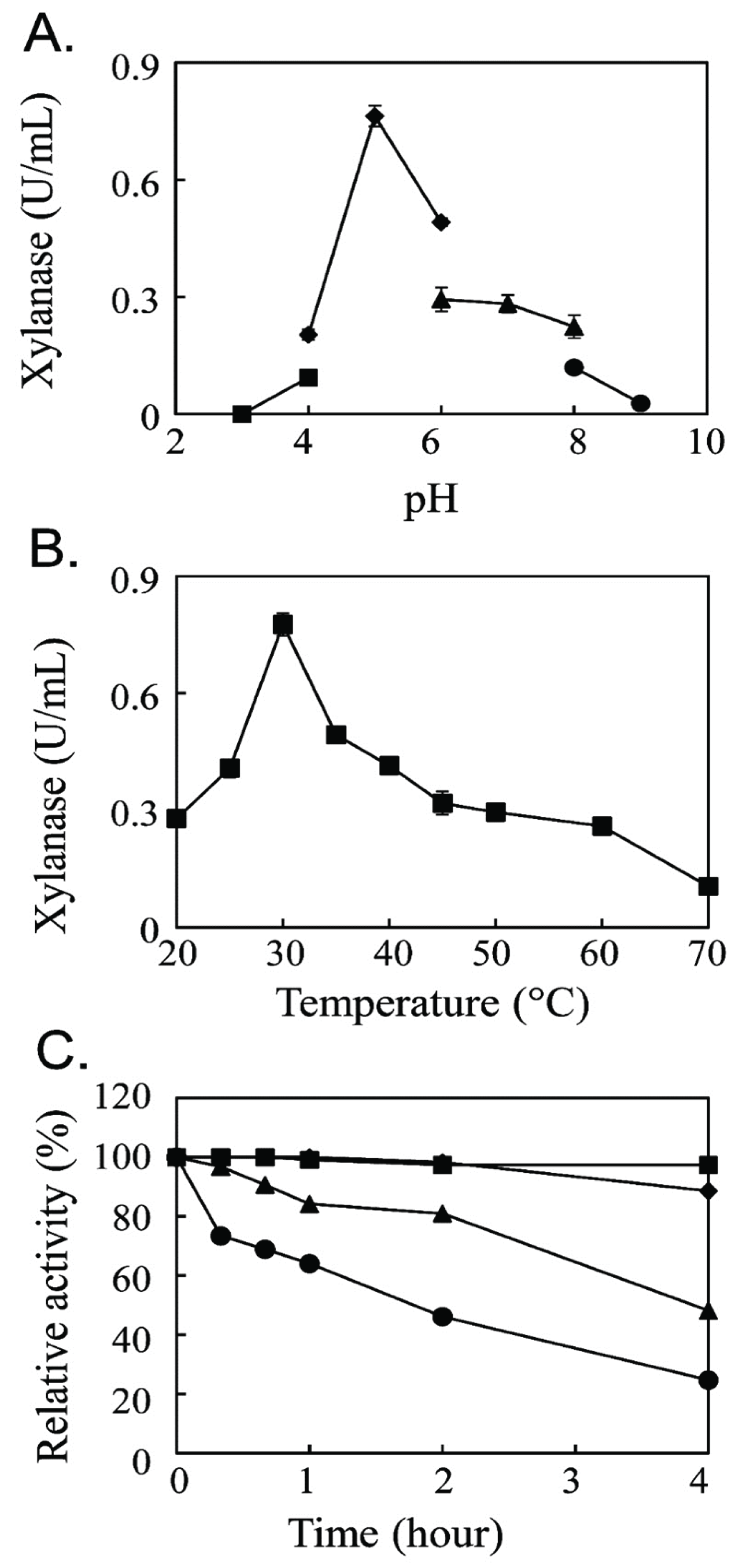
The optimal reaction condition for the new endo-β-1,4-xylanase was determined (Fig. 2). The xylanase activity was maximum at pH 5.0 with 100 mM sodium acetate buffer (Fig. 2A) and at 30°C (Fig. 2B). The optimum reaction at pH 5.0 was different from that of the bacterial endo-β-1,4-xylanase with the optimum reaction at pH 7.0 (Guo et al., 2009) and pH 9.0 (Khandeparkar and Bhosle, 2006) but was similar to the optimum reaction pH of the fungal xylanase from Aspergillus spp. (Wong et al., 1988). The optimum reaction temperature of the novel endo-β-1,4-xylanase at 30°C showed that it was a mesophilic enzyme. The thermal stability analysis showed that the enzymatic activity of the new xylanase was decreased by half at 30°C for 4 h (Fig. 2C).
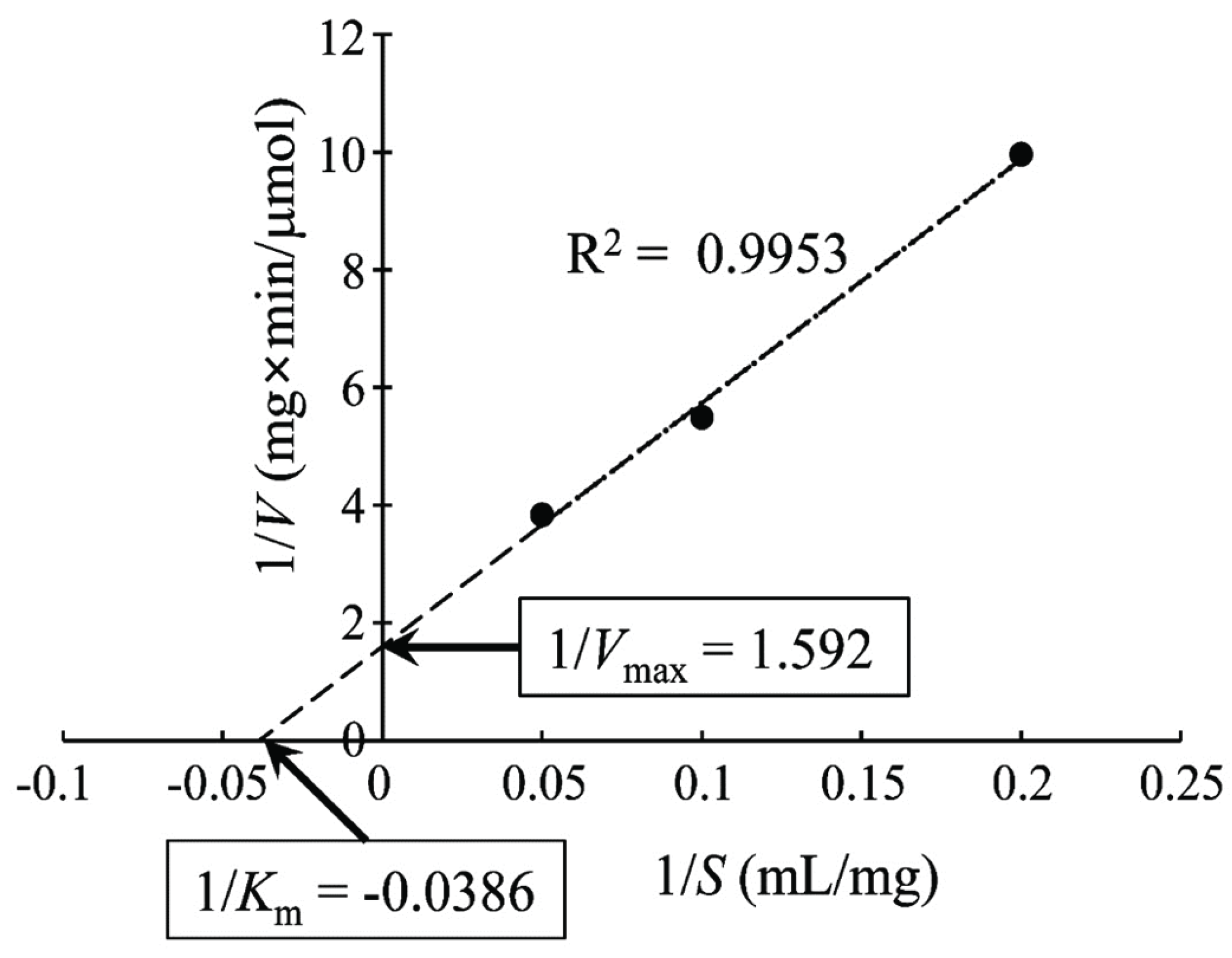
To determine the Km and Vmax of the new xylanase, the Lineweaver–Burk plot was drawn, as shown in Fig. 3. The kinetic parameter Km of the novel xylanase was 25.92 mg/ml, which is similar to that of xylanase from Aspergillus niger (Liu et al., 2011) but higher than that of xylanases from Streptomyces sp. (Wang et al., 2014) and a fungus, Orpinomyces sp. (Trevizano et al., 2012). This high Km value indicated a strong dependency of the novel xylanase on the substrate concentration. The enzyme kinetic value of Vmax was 0.628 μmole/mg/ml.
Because some xylanase activity was increased by ions (Khandeparkar and Bhosle, 2006; Guo et al., 2009), the change in the new endo-β-1,4-xylanase activity due to the ions was measured (Table 3). Only calcium ion increased the enzyme activity by 126% and such an increased xylanase activity by calcium has also been demonstrated in bacteria (Spurway et al., 1997; Yazawa et al., 2011). Calcium ion increased the enzymatic activity of several glycoside hydrolases (Okuyama et al., 2014). The thermostable xylanase activity from Arthrobacter sp. increased with Ca+2 (Khandeparkar and Bhosle, 2006). All the observed characteristics of the novel xylanase indicated a typical fungal xylanase.
| Metal ions (1 mM) | Relative enzyme activity (%) |
|---|---|
| Control | 100 |
| Cu+2 | N.D. |
| Ca+2 | 226 |
| Co+2 | N.D. |
| K+ | 53 |
| Ba+2 | 23 |
| Fe+2 | 2 |
| Zn+2 | 106 |
| Mn+2 | N.D. |
| Mg+2 | N.D. |
| Fe3+ | N.D. |
| Na+ | 21 |
4. CONCLUSION
In this study, it was identified that the new protein, encoded by isotig01043 and expressed heterogeneously in E. coli BL21(DE3), exhibited a xylanase activity. A low identity (83%) with the closest protein in the NCBI database and the unreported xylanase activity of a similar domain with GH16_fungal_Lam16A_ glucanase domain suggested that the protein encoded by isotig01043 was a new type of xylanase.